
Learn more about the current most used compounds in research with the #Wikimole campaign!
Contents
A 83-01
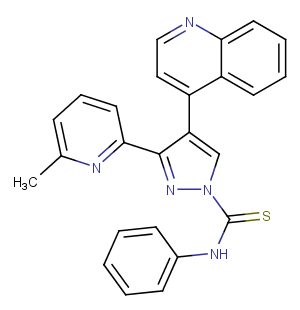
A 83-01, also known as ALK5 Inhibitor IV, is an inhibitor of TGF-β type I receptors ALK5, ALK4, and ALK7 (IC50 = 12/45/7.5 nM). A 83-01 promotes the reprogramming of mouse fibroblasts into induced pluripotent stem cells (iPSCs). It can also be used for organoid culture.
Bioactivity: A 83-01 is a selective inhibitor of TGF-β type I receptor ALK5 kinase, type I activin/nodal receptor ALK4 and type I nodal receptor ALK7 with IC50 values of 12 nM, 45 nM and 7.5 nM, respectively.
Biological Applications: There are many small molecules used in reprogramming, which can be categorised into 4 types: metabolic regulators, epigenetic modifiers, signalling modulators, and ageing inhibitors.
A 83-01 inhibits the activity of TGF-β type I receptors ALK5/4/7 kinases. TGF-β plays a crucial role in stem cell culture. Stem cells possess active paracrine functions and can secrete a large amount of transforming growth factors through exosomes. TGF-β promotes stem cell proliferation, aiding in the maintenance of their numbers. A 83-01, as a TGF-β inhibitor, is commonly used to inhibit the differentiation of iPSCs and maintain the self-renewal of cells in vitro. A 83-01 is generally used for the culture of gastrointestinal, hepatic, prostatic, and mammary organoids.
Research Area: Organoid
Target: ALK; TGF-beta/Smad
BI-2493
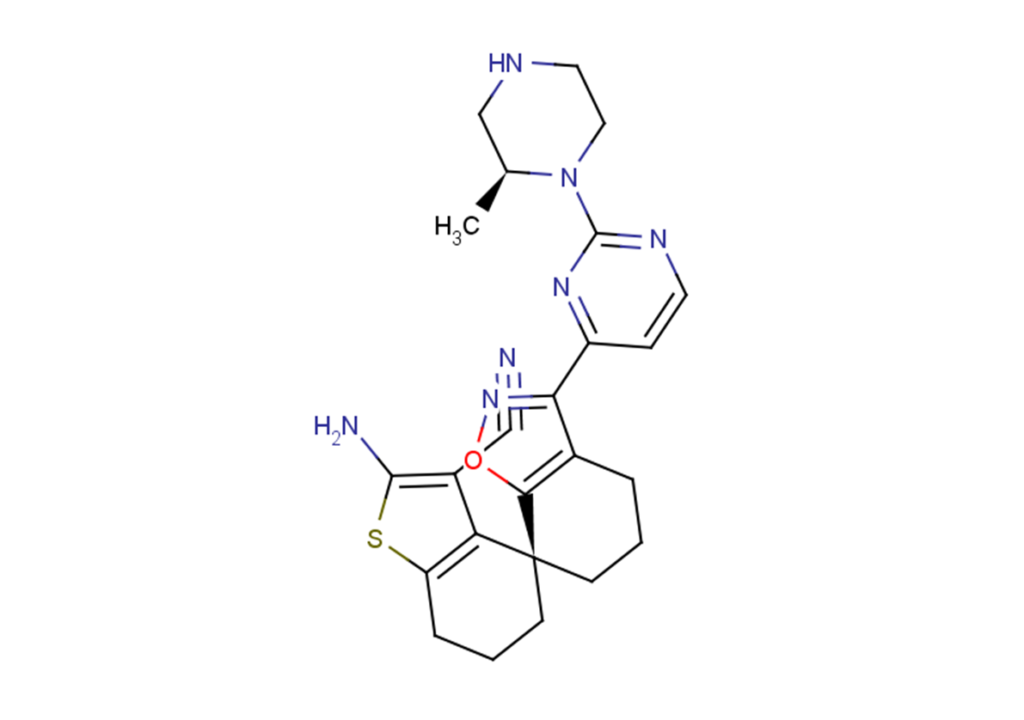
BI-2493 is a highly selective pan-KRAS inhibitor and a structural analogue of BI-2865. It exhibits similar anti-tumour activity to BI-2865, inhibiting tumour cell growth, and is used in research related to cancer diseases.
Bioactivity: BI-2493 is a highly selective pan-KRAS inhibitor and structural analogue of BI-2865. BI-2493 exhibits anti-tumour activity, inhibits tumour cell growth and can be used in the study of cancerous diseases.
Biological Applications: Pan-KRAS Inhibitors – More than 100 mutation sites have been identified in RAS subtypes, with prominent mutation hotspots at G12, G13, and Q61. However, KRAS-specific inhibitor targets only G12C or G12D mutants, limiting clinical applications. To address these limitations, research teams have begun developing small-molecule pan-KRAS inhibitors.
Pan-KRAS inhibitors deactivate common KRAS cancer proteins without the need for covalent anchoring to specific mutant amino acids. These inhibitors can prevent reactivation through nucleotide exchange by preferentially targeting the non-active state of KRAS. Representative examples include BI-2865 and its analogue BI-2493.
Research Area: Pan-KRAS Inhibitors
Target: Ras
BI-2865
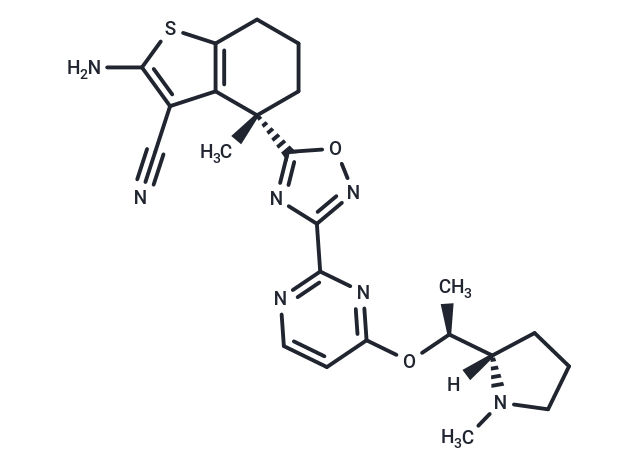
BI-2865 is a non-covalent inhibitor with pan-KRAS potential. This inhibitor induces the inactivation of common KRAS oncoproteins (BI-2865 targets KRAS WT, G12C, G12D, G12V, and G13D mutants) without the need for covalent anchoring to specific mutant amino acids.
Bioactivity: BI-2865 is a non-covalent pan-KRAS inhibitor. BI-2865 binds to KRAS WT, G12C, G12D, G12V, and G13D mutants with KD values of 6.9, 4.5, 32, 26, and 4.3 nM, respectively. BI-2865 showed antiproliferative activity in BaF3 cells expressing KRAS G12C, G12D, or G12V mutants. BI-2865 showed antiproliferative activity on BaF3 cells expressing KRAS G12C, G12D or G12V mutants with an average IC50 value of approximately 140 nM.
Biological Applications:
Pan-KRAS Inhibitors – More than 100 mutation sites have been identified in RAS subtypes, with prominent mutation hotspots at G12, G13, and Q61. However, KRAS-specific inhibitor targets only G12C or G12D mutants, limiting clinical applications. To address these limitations, research teams have begun developing small-molecule pan-KRAS inhibitors.
Pan-KRAS inhibitors deactivate common KRAS cancer proteins without the need for covalent anchoring to specific mutant amino acids. These inhibitors can prevent reactivation through nucleotide exchange by preferentially targeting the non-active state of KRAS. Representative examples include BI-2865 and its analogue BI-2493.
Research Area: Pan-KRAS Inhibitors
Target: Ras
CHIR-99021
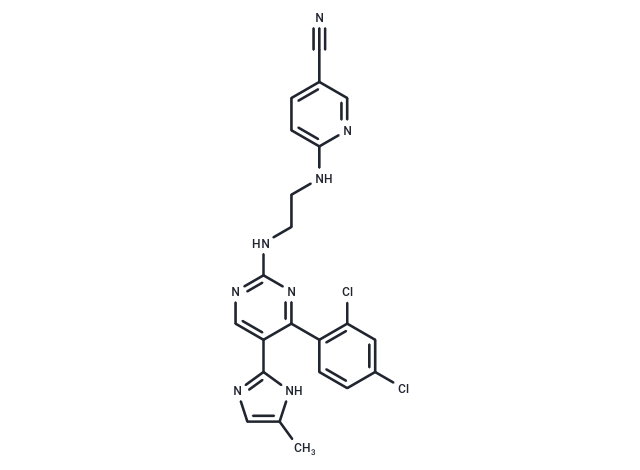
CHIR-99021, also known as Laduviglusib or CT99021, is a highly selective inhibitor of GSK-3α/β (Glycogen synthase kinase 3), with IC50 values of 10 nM and 6.7 nM, respectively. It is also an effective activator of the Wnt/β-catenin signalling pathway. Additionally, CHIR-99021 can induce autophagy and enhance self-renewal in mouse and human embryonic stem cells.
Bioactivity: CHIR-99021 is a GSK-3α/β inhibitor (IC50: 10/6.7 nM).
Biological Applications: CHIR-99021 is a highly selective inhibitor of GSK-3α/β, regulators of the Wnt signalling pathway. By inhibiting GSK-3α/β, CHIR-99021 promotes the activation of the Wnt signalling pathway, thereby influencing the self-renewal and differentiation of stem cells. For example, human pluripotent stem cell (hPSC) -derived organoids such as Heart Forming Organoids (HFOs) represent a complex, highly structured in vitro model of early heart, gut, and vascular system development. These organoids are commonly used in teratogenic research, gene function analysis, and drug discovery studies. CHIR-99021 and IWP2 (Catalog No. T2702) are typically used during the establishment of this model for cell differentiation. As research progresses, scientists have found that CHIR-99021 significantly enhances the quantity and functionality of stem cells, showing promising applications across various stem cell types. The scope of CHIR-99021’s applications continues to expand. Apart from its use in the field of stem cells, CHIR-99021 also demonstrates potential therapeutic value in neurodegenerative diseases, tumours, and metabolic disorders. In neurodegenerative diseases, CHIR-99021 can inhibit the generation and aggregation of beta-amyloid proteins, thereby slowing the progression of conditions like Alzheimer’s disease. In the field of tumours, CHIR-99021 can modulate tumour cell proliferation and apoptosis, inhibiting tumour growth and metastasis. Additionally, CHIR-99021 can improve symptoms of metabolic disorders such as diabetes and obesity, promoting metabolic balance in the body.
Research Area: Type 2 diabetes, Alzheimer’s disease, inflammation, cancer, addiction, and bipolar disorder.
Target: GSK-3; Wnt/beta-catenin; Autophagy
CHIR-99021 HCl
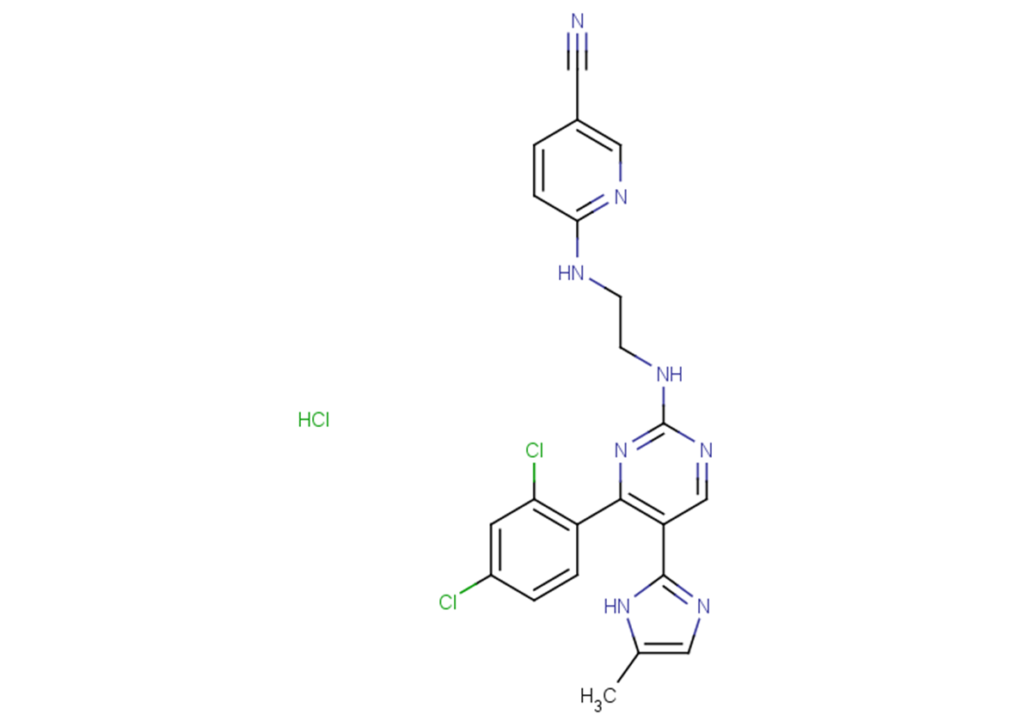
CHIR-99021 HCl is the hydrochloride salt form of CHIR-99021.
Bioactivity: Laduviglusib (CHIR-99021) monohydrochloride is a highly potent and selective inhibitor of GSK-3α/β, with IC50 values of 10 nM and 6.7 nM respectively. It demonstrates remarkable selectivity for GSK-3, with over 500-fold selectivity over CDC2, ERK2, and other protein kinases. Additionally, Laduviglusib monohydrochloride serves as a robust activator of the Wnt/β-catenin signalling pathway. Moreover, it exhibits the ability to enhance self-renewal in both mouse and human embryonic stem cells. Furthermore, Laduviglusib monohydrochloride induces autophagy.
Biological Applications: CHIR-99021 is a highly selective inhibitor of GSK-3α/β, which are regulators of the Wnt signalling pathway. By inhibiting GSK-3α/β, CHIR-99021 promotes the activation of the Wnt signalling pathway, thereby influencing the self-renewal and differentiation of stem cells. For example, human pluripotent stem cell (hPSC)-derived organoids such as Heart Forming Organoids (HFOs) represent a complex, highly structured in vitro model of early heart, gut, and vascular system development. These organoids are commonly used in teratogenic research, gene function analysis, and drug discovery studies. CHIR-99021 and IWP2 (Catalog No. T2702) are typically used during the establishment of this model for cell differentiation. As research progresses, scientists have found that CHIR-99021 significantly enhances the quantity and functionality of stem cells, showing promising applications across various stem cell types. The scope of CHIR-99021’s applications continues to expand. Apart from its use in the field of stem cells, CHIR-99021 also demonstrates potential therapeutic value in neurodegenerative diseases, tumours, and metabolic disorders. In neurodegenerative diseases, CHIR-99021 can inhibit the generation and aggregation of beta-amyloid proteins, thereby slowing the progression of conditions like Alzheimer’s disease. In the field of tumours, CHIR-99021 can modulate tumour cell proliferation and apoptosis, inhibiting tumour growth and metastasis. Additionally, CHIR-99021 can improve symptoms of metabolic disorders such as diabetes and obesity, promoting metabolic balance in the body.
Research Area: Type 2 diabetes, Alzheimer’s disease, inflammation, cancer, addiction, and bipolar disorder.
Target: GSK-3
Durlobactam sodium salt
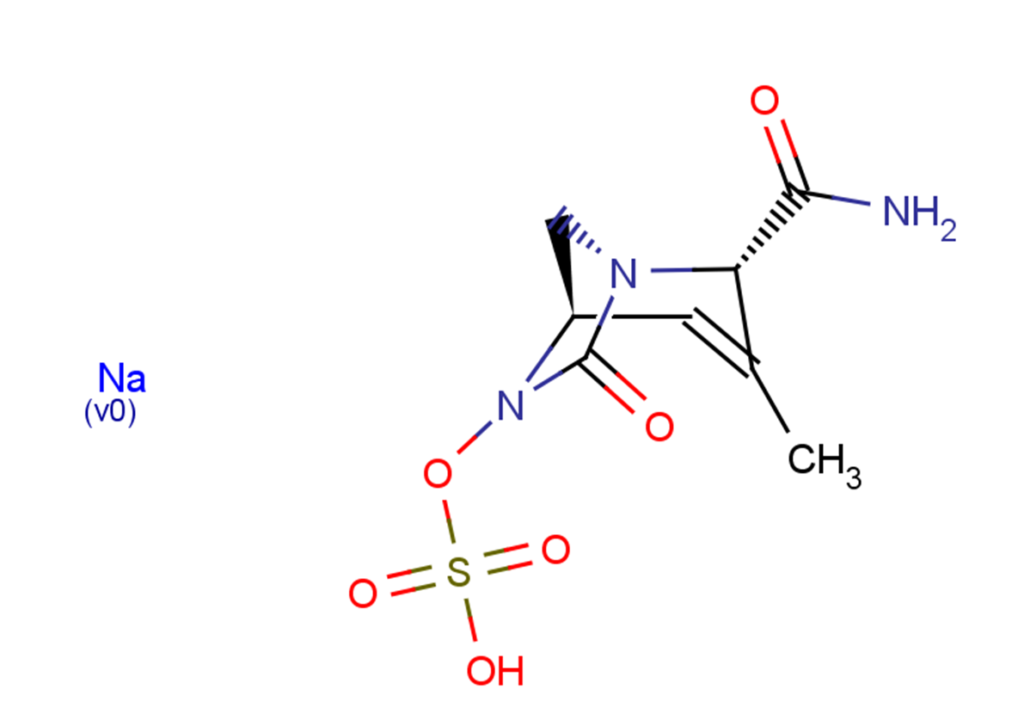
Durlobactam sodium salt is the sodium salt form of Durlobactam, also known as ETX2514, Duobatan sodium, and Durobatan sodium. It is a beta-lactamase inhibitor with varying degrees of inhibition against beta-lactamases of classes A, C, and D. Durlobactam sodium salt can be used for research on multidrug-resistant Gram-negative bacteria, including Acinetobacter baumannii.
Bioactivity: Durlobactam sodium salt is an inhibitor of β-lactamase with IC50 values of 4 nM, 14 nM, and 190 nM for Class A KPC-2, Class C AmpC, and Class D OXA-24, respectively. Durlobactam sodium salt can be used in studies about drug-resistant Gram-negative bacteria including Acinetobacter baumannii.
Biological Applications: Durlobactam is a novel broad-spectrum intravenous beta-lactamase inhibitor commonly used in combination with beta-lactam antibiotics, particularly Sulbactam. Sulbactam is an intravenous beta-lactam antibiotic with intrinsic antibacterial activity against Acinetobacter baumannii complex (ABC) infections. In May 2023, Sulbactam/Durlobactam (XACDURO®) received FDA approval in the United States for patients aged 18 and older, indicated for the treatment of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia (HABP/VABP) caused by susceptible strains of ABC. This medication is a combination product designed to address infections caused by the Acinetobacter baumannii-Calcoaceticus-Baumannii complex. The use of Durlobactam prevents the degradation of Sulbactam by beta-lactamases produced by ABC, enhancing its efficacy.
Mechanism Of Action: Beta-lactamases are enzymes produced by certain bacteria, categorised into four classes (A, B, C, and D). These enzymes contribute to bacterial multidrug resistance by inactivating beta-lactam antibiotics, such as penicillins, cephalosporins, monobactams, and carbapenems. Beta-lactam antibiotics constitute a widely used class of antibiotics characterised by a common four-membered core structure known as a beta-lactam ring. Beta-lactamases hydrolyse the beta-lactam ring, rendering the antibiotics ineffective and leading to bacterial resistance.
Gram-negative bacteria commonly produce beta-lactamases, and clinically relevant organisms include members of the Acinetobacter–Calcoaceticus–Baumannii (ACB) complex. The rapid acquisition of multidrug resistance, encompassing fluoroquinolones, aminoglycosides, cephalosporins, and carbapenems, by the ACB complex limits therapeutic options, posing a significant global public health threat. The development of drugs targeting such bacteria is of crucial clinical importance.
Research Area: Gram-negative bacteria study
Target: Antibacterial
Eragidomide
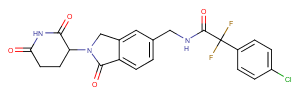
Eragidomide, also known as CC-90009 or Cereblon modulator 1, is a selective cereblon (CRBN) E3 ubiquitin ligase modulator with specificity towards GSPT1. It acts through molecular glue, selectively targeting GSPT1 for ubiquitination and proteasomal degradation via the CRL4CRBN pathway.
Bioactivity: Cereblon modulator 1 is a cereblon (CRBN) E3 ligase modulator. It specifically binds to CRBN, thereby affecting the activity of the ubiquitin E3 ligase complex.
Biological Applications: The human body encodes over 600 different E3 ubiquitin ligases, but currently, only about 10 publicly disclosed E3 ligases are used in protein degradation research. Among them, two E3 ligases, CRBN and VHL12, have entered the clinical stage for protein degradation studies. Eragidomide (CC-90009) and Mezigdomide (CC-92480) are both CRBN E3 ligase modulators (CELMoD), exhibiting potent anti-tumour and immunomodulatory effects. They particularly induce immune-stimulatory effects and enhanced anti-tumour activity against multiple myeloma cells. They are currently undergoing clinical trials as potential treatments for diseases such as multiple myeloma. CELMoD stands for Cereblon E3 Ligase Modulator, and it is a small molecule that can bind to E3 ligases (such as CRBN). Subsequently, it recruits new substrate proteins for ubiquitination and proteasomal degradation. GSPT1, also known as G1 to S phase transition 1, is a crucial translation termination factor significantly overexpressed in various cancer tissues and cells. Molecular glue refers to a small molecule that can bind to E3 ligases (e.g., CRBN) and then recruit new substrate proteins for ubiquitination and proteasomal degradation.
Mechanism of Action: E3 ubiquitin ligases play a crucial role in protein degradation. Ubiquitin (Ub) is a small protein consisting of approximately 76 amino acids with a molecular weight of around 8.5 kDa, and it is widely present in all eukaryotic cells. The process in which ubiquitin is covalently attached to target proteins, under the catalytic action of a series of enzymes, is known as ubiquitination. This is a highly regulated post-translational modification of proteins that not only participates in protein degradation but also plays a vital role in regulating cellular functions. Ubiquitination typically involves the coordinated action of three enzymes: E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme, and E3 ubiquitin ligase. The process is:
- Ubiquitin-activating enzyme (E1) activates the ubiquitin molecule using ATP as an energy source.
- E1 transfers the activated ubiquitin molecule to the ubiquitin-conjugating enzyme (E2).
- Ubiquitin ligase (E3) facilitates the transfer of ubiquitin from the E2 enzyme to a specific lysine residue on the target protein.
In summary, the coordinated action of ubiquitin ligases allows ubiquitin molecules to be covalently attached to target proteins, thereby regulating the function, stability, and localisation of proteins. E3 ubiquitin ligases, as a critical final step, hold significant importance in the field of protein degradation and provide powerful tools for the development of novel drugs.
Research Area: Haematological Oncology, Prostate Cancer, Immune Microenvironment, and New drug discovery targeting protein degradation.
Target: Apoptosis
Gastrin I, human
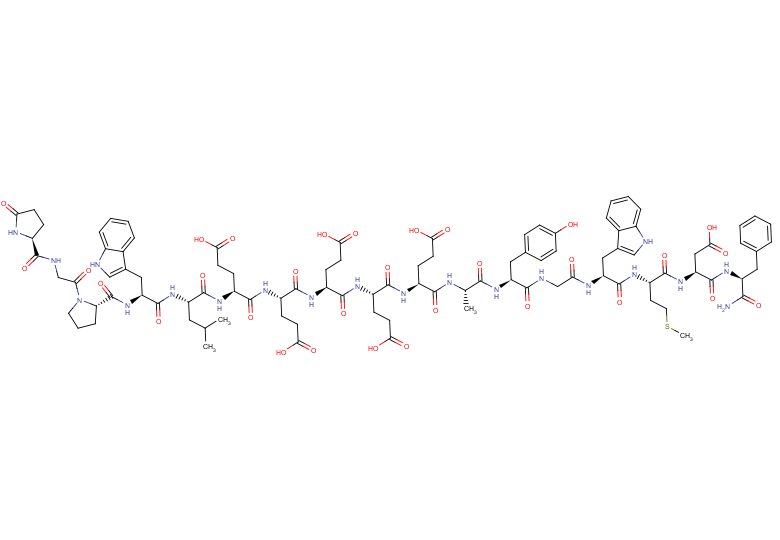
Gastrin I, human, also known as Gastrin I (human) or Gastrin 1, is an endogenous peptide produced in the stomach. It increases gastric pepsinogen and gastric acid secretion in rats through the CCK2 receptor.
Bioactivity: Endogenous peptide produced in the stomach that acts as a selective CCK2 receptor agonist and stimulates gastric acid secretion. It is also used in the culture of stomach organoids.
Biological Applications: In the culture process of gastrointestinal organs, gastrin I can promote gastric acid secretion, proliferation, and repair of gastric mucosal cells. It also simulates the physiological environment in the body, helping to maintain the normal development and function of gastric organs. Therefore, in the culture process of gastrointestinal organs, gastrin I is often added as an auxiliary factor to the culture medium.
Mechanism of Action: Gastrin is an important gastrointestinal hormone mainly secreted by G cells. G cells are typical open-type cells, most abundant in the gastric antrum, followed by the gastric fundus, duodenum, and jejunum. Gastrin I is one of the earliest discovered subtypes of gastrin. Gastrin affects almost the entire gastrointestinal tract. It can promote the synthesis of DNA, RNA, and proteins in the mucosa of the gastric acid gland area and duodenal mucosa, thereby promoting mucosal cell growth and proliferation. The main functions of gastrin include:
- Stimulating the synthesis of DNA, RNA, and proteins in the mucosa of the gastric acid gland area and duodenal mucosa, thereby promoting mucosal cell growth and proliferation.
- Stimulating parietal cells to secrete hydrochloric acid and chief cells to secrete pepsinogen.
- Stimulating gastric antrum and intestinal movement, delaying gastric emptying.
- Stimulating the secretion of pancreatic juice, bile, and intestinal juice.
Research Area: Organiod (gastrointestinal, liver, pancreas)
Target: Cholecystokinin
Mezigdomide
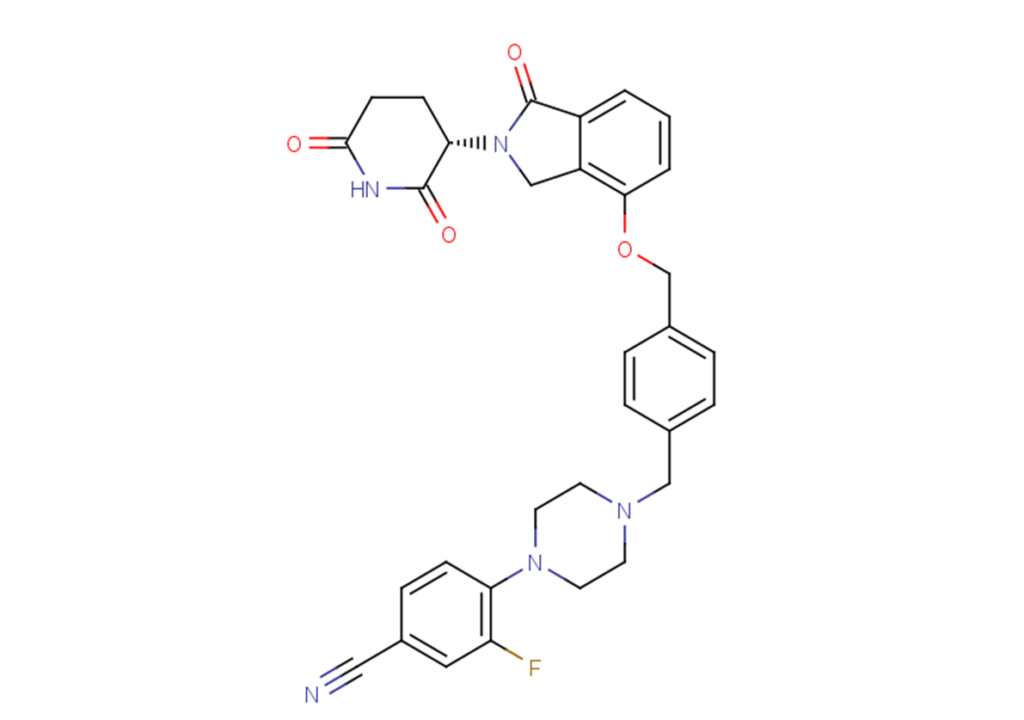
Mezigdomide, alias CC-92480, is a novel and selective CRBN E3 ubiquitin ligase modulator. It functions in the form of a molecular glue to recruit IKZF1 and ZFP91 targets to the CRL4CRBN E3 ubiquitin ligase, leading to their ubiquitination and degradation.
Bioactivity: CC-92480 is a cereblon E3 ubiquitin ligase modulating drug (CELMoD) with potent antimyeloma activity.
Biological Applications: The human body encodes over 600 different E3 ubiquitin ligases, but currently, only about 10 publicly disclosed E3 ligases are used in protein degradation research. Among them, two E3 ligases, CRBN and VHL12, have entered the clinical stage for protein degradation studies. Eragidomide (CC-90009) and Mezigdomide (CC-92480) are both CRBN E3 ligase modulators (CELMoD), exhibiting potent anti-tumour and immunomodulatory effects. They particularly induce immune-stimulatory effects and enhanced anti-tumour activity against multiple myeloma cells. They are currently undergoing clinical trials as potential treatments for diseases such as multiple myeloma. CELMoD stands for Cereblon E3 Ligase Modulator, and it is a small molecule that can bind to E3 ligases (such as CRBN). Subsequently, it recruits new substrate proteins for ubiquitination and proteasomal degradation. GSPT1, also known as G1 to S phase transition 1, is a crucial translation termination factor significantly overexpressed in various cancer tissues and cells. Molecular glue refers to a small molecule that can bind to E3 ligases (e.g., CRBN) and then recruit new substrate proteins for ubiquitination and proteasomal degradation.
Mechanism of Action: E3 ubiquitin ligases play a crucial role in protein degradation. Ubiquitin (Ub) is a small protein consisting of approximately 76 amino acids with a molecular weight of around 8.5 kDa, and it is widely present in all eukaryotic cells. The process in which ubiquitin is covalently attached to target proteins, under the catalytic action of a series of enzymes, is known as ubiquitination. This is a highly regulated post-translational modification of proteins that not only participates in protein degradation but also plays a vital role in regulating cellular functions. Ubiquitination typically involves the coordinated action of three enzymes: E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme, and E3 ubiquitin ligase. The process is:
- Ubiquitin-activating enzyme (E1) activates the ubiquitin molecule using ATP as an energy source.
- E1 transfers the activated ubiquitin molecule to the ubiquitin-conjugating enzyme (E2).
- Ubiquitin ligase (E3) facilitates the transfer of ubiquitin from the E2 enzyme to a specific lysine residue on the target protein.
In summary, the coordinated action of ubiquitin ligases allows ubiquitin molecules to be covalently attached to target proteins, thereby regulating the function, stability, and localisation of proteins. E3 ubiquitin ligases, as a critical final step, hold significant importance in the field of protein degradation and provide powerful tools for the development of novel drugs.
Research Area: Haematological Oncology, Prostate Cancer, Immune Microenvironment, and New drug discovery targeting protein degradation.
Target: Apoptosis; E1/E2/E3 Enzyme
MG-132
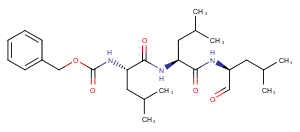
MG-132, also known as Z-LLL-al or Z-Leu-Leu-Leu-CHO, is a 26S proteasome inhibitor with an IC50 of 100 nM. It exhibits cell permeability and reversibility. MG-132 is capable of inducing apoptosis.
Bioactivity: MG-132 is a potent cell-permeable 26S proteasome inhibitor (IC50: 100 nM). It also inhibits calpain (IC50: 1.2 μM).
Biological Applications: The 26S proteasome maintains cellular protein homeostasis by clearing abnormal, aging, or excessive proteins. It participates in the regulation of cellular life cycles, stress responses, and metabolic regulation, among other physiological processes. MG-132, as an inhibitor of the 26S proteasome, can disrupt the normal function of this system, providing important tools and information for research in cell biology and molecular biology. Additionally, the apoptosis of cancer cells is closely related to the activity of the ubiquitin-proteasome pathway. MG-132 can induce cell apoptosis through various intermediate pathways, playing a crucial role in anti-tumour therapy.
Mechanism of Action: The 26S proteasome is one of the most important protein degradation systems in cells, also known as the Ubiquitin-Proteasome System (UPS). This system mainly consists of two parts: the ubiquitination system and the proteasome.
Ubiquitination system: This system involves the covalent attachment of ubiquitin molecules to target proteins, marking these proteins as targets for degradation. This process involves ubiquitin-activating enzymes and ubiquitin-conjugating enzymes, which work together to attach ubiquitin to specific proteins, forming polyubiquitin chains.
Proteasome: This system is mainly composed of 20S core particles and 19S regulatory particles that bind to them, forming a 26S complex. The 26S proteasome is the primary intracellular protease responsible for degrading proteins that have been tagged with ubiquitin. It plays a crucial role in maintaining the quality of intracellular proteins and regulating protein levels by recognising, deconstructing, and degrading ubiquitinated proteins.
Research Area: Epigenetics, regulation of gene expression and cell division, proliferation, and molecular biology.
Target: Apoptosis; Proteasome; Autophagy
Olaparib
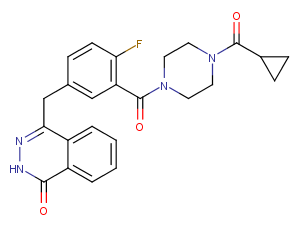
Olaparib, also known as AZD2281 or KU0059436, is a small molecule inhibitor of PARP1/PARP2. It exhibits selectivity and oral activity. Additionally, Olaparib possesses activity in inducing autophagy and mitochondrial autophagy.
Bioactivity: Olaparib is a small molecule inhibitor of PARP1/PARP2 (IC50: 5/1 nM) but is less effective against the PARP tankyrase-1 (IC50: 1.5 μM).
Biological Applications: Olaparib, marketed under the brand name Lynparza®, is an approved PARP inhibitor used to treat ovarian cancer and breast cancer patients carrying BRCA mutations. For these patients, specific gene mutations like BRCA disrupt other DNA repair pathways, making them particularly sensitive to PARP inhibitors. Treating ovarian and breast cancer patients carrying BRCA mutations is a common application of PARP inhibitors. Additionally, PARP inhibitors have shown therapeutic potential in other cancer types and non-oncological diseases. Approved PARP inhibitors on the market include Olaparib, Rucaparib, Niraparib, Talazoparib, etc.
Research Area: Breast cancer and Ovarian cancer.
Target: Mitophagy; PARP; Autophagy
Orforglipron
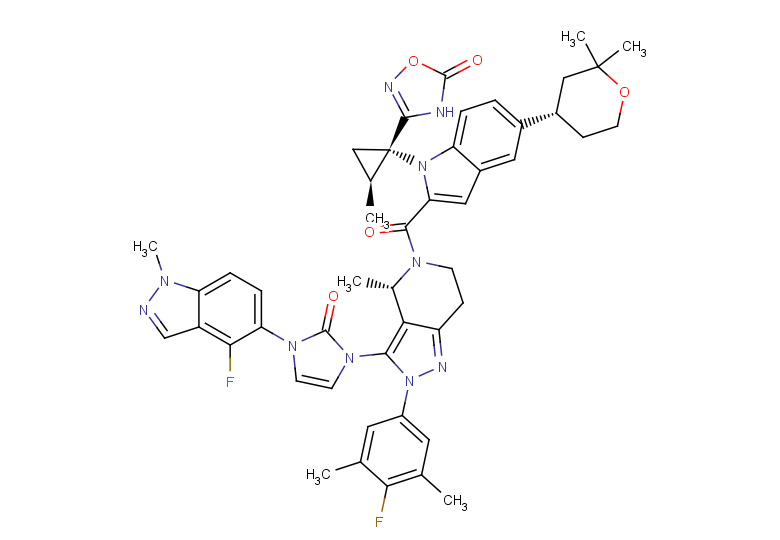
GLP-1RA and SGLT-2i are new diabetes medications that have gained widespread attention due to their cardiovascular and renal benefits brought to patients with type 2 diabetes (T2D). Orforglipron, also known as LY3502970, is a GLP-1 receptor agonist. It is used in research related to obesity and T2D.
Bioactivity: GLP-1 receptor agonist 1 is a GLP-1 receptor agonist .
Research Area: Obesity and Type 2 Diabetes.
Target: Glucagon Receptor
SB 202190
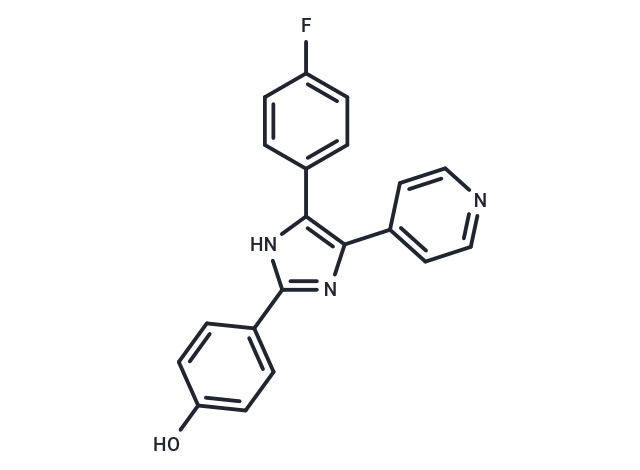
SB 202190, also known as FHPI, is a selective inhibitor of p38 MAPK. It inhibits p38α and p38β2 with IC50 values of 50 nM and 100 nM, respectively. Additionally, it has demonstrated efficacy in rescuing memory impairments and exhibits anticancer activity. SB 202190 can also be used in organoid culture.
Bioactivity: SB 202190 is a selective and cell-permeable inhibitor of p38 MAPK (IC50s: 50/100 nM for p38α/p38β).
Biological Applications: There are many small molecules used in reprogramming, which can be categorised into four types: metabolic regulators, epigenetic modifiers, signalling modulators, and ageing inhibitors.
SB 202190 is a potent p38 MAPK kinase inhibitor that can induce human embryonic stem cells to differentiate into cardiac muscle cells and promote the self-renewal of neural stem cells. It can be used for the culture of gastrointestinal, mammary, and prostatic organoids.
Research Area: Organoid
Target: Apoptosis; p38 MAPK; Autophagy
SB-431542
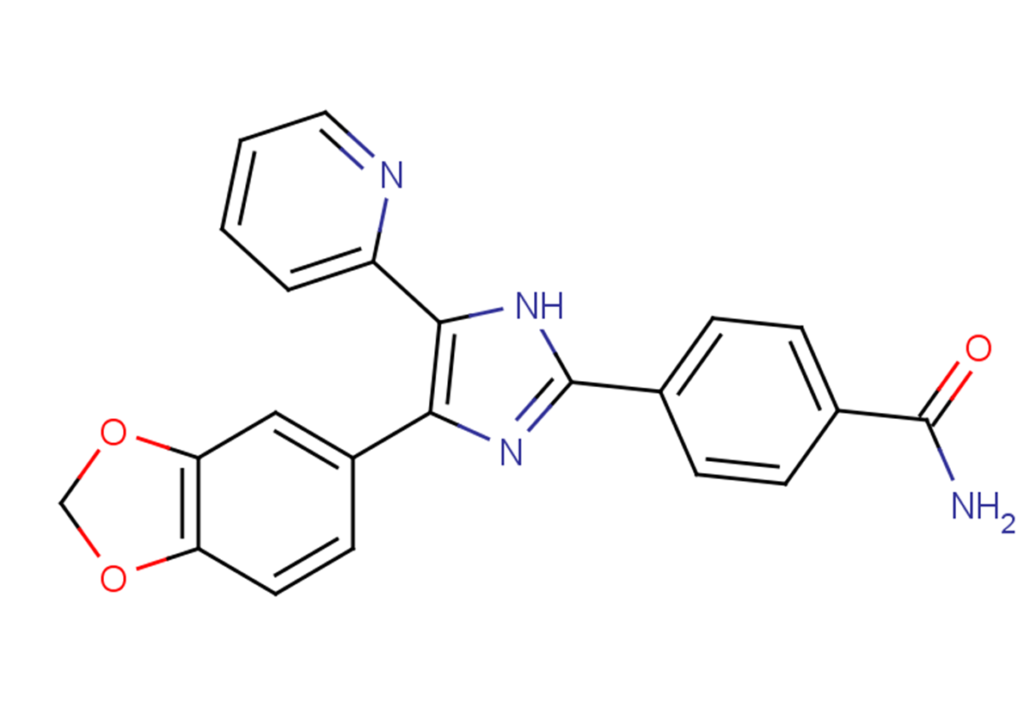
SB-431542, also known as SB 431542 or 4-[4-(1,3-benzodioxol-5-yl)-5-(2-pyridinyl)-1H-imidazol-2-yl]benzamide hydrate, is a selective inhibitor of transforming growth factor-beta (TGF-β) type I receptor ALK5, with an IC50 of 94 nM. It also exhibits inhibitory activity against ALK4 and ALK7 to some extent, with no inhibitory effect on other proteins. SB-431542 is commonly used for inducing differentiation in stem cells.
Bioactivity: SB-431542 is a potent and selective inhibitor of ALK5 (IC50: 94 nM) and is also an inhibitor of ALK4 (IC50: 140 nM) and ALK7.
Biological Applications: There are many small molecules used in reprogramming, which can be categorized into four types: metabolic regulators, epigenetic modifiers, signalling modulators, and ageing inhibitors.
SB-431542 inhibits the activity of TGF-β type I receptors ALK5/4/7 kinases. TGF-β plays a crucial role in stem cell culture. Stem cells possess active paracrine functions and can secrete a large amount of transforming growth factors through exosomes. TGF-β promotes stem cell proliferation, aiding in the maintenance of their numbers.SB-431542, as a TGF-β inhibitor, is commonly used to inhibit the differentiation of iPSCs and maintain the self-renewal of cells in vitro. SB-431542 is typically used for the culture of lung and inner ear organoids.
Research Area: Organoid
Target: ALK; TGF-beta/Smad
Y-27632
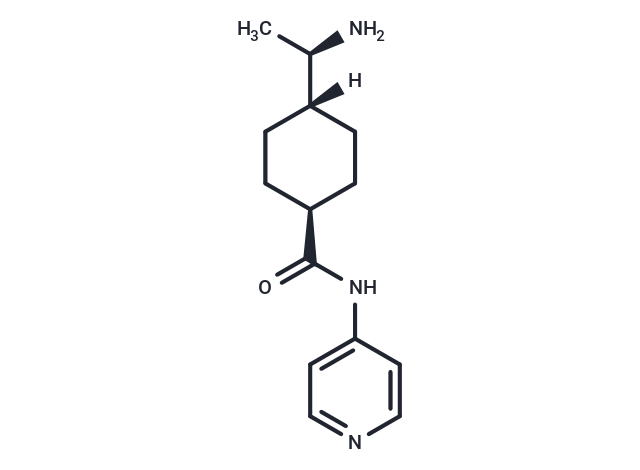
Y-27632 is a selective inhibitor of ROCK-I and ROCK-II, exhibiting oral bioavailability and ATP competitiveness. Y-27632 can also inhibit apoptosis of dissociation-induced mouse prostate stem cells or progenitor cells, commonly used in stem cell research and organoid culture.
Bioactivity: Y-27632 is a selective ATP-competitive inhibitor of ROCK-I and ROCK-II.
Biological Applications: Y-27632, as a ROCK inhibitor, has significant applications not only in diseases like cancer but also in organoid culture and stem cell research, contributing to the maintenance of cell survival and functionality, thus holding crucial value in various fields. Common applications:
- Cryopreservation of stem cells: Y-27632 helps prevent dissociation-related cell apoptosis during the low-temperature preservation of stem cells and enhances cell viability post-thaw.
- Pancreatic ductal adenocarcinoma organoid culture: Y-27632 serves as a supplement to the culture medium.
- Mouse embryonic stem cells: Y-27632 inhibits the Rho kinase (Rho) in these cells.
- Human embryonic stem cells and induced pluripotent stem cells (iPSCs): Y-27632 inhibits Rho-associated protein kinase (ROCK).
These diverse applications highlight the versatility and importance of Y-27632 in various research settings, promising advancements in cell biology and regenerative medicine.
Research Area: Cancer, glaucoma, asthma, erectile dysfunction, insulin resistance, neurodegeneration, osteoporosis, renal failure, fibrosis, and graft-versus-host disease.
Target: ROCK; Apoptosis
Y-27632 dihydrochloride
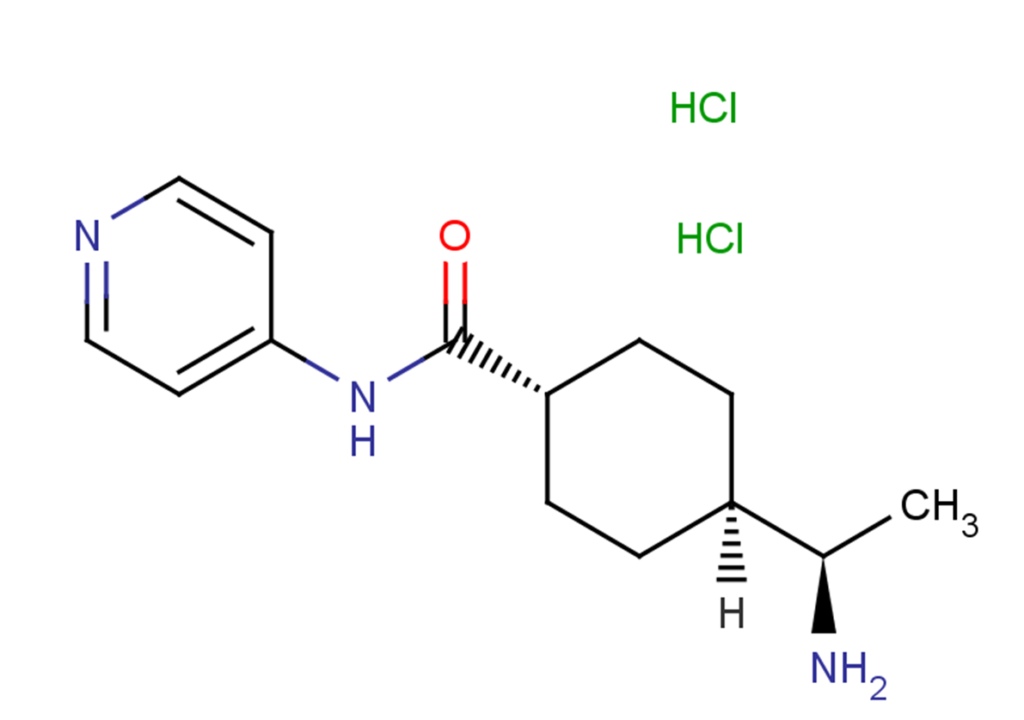
Y-27632 2HCl, also known as Y-27632 dihydrochloride or trans-4-[(R)-1-aminoethyl]-N-(4-pyridyl)cyclohexanecarboxamide dihydrochloride, is the dihydrochloride salt form of Y-27632, exerting the same inhibitory effects as Y-27632.
Bioactivity: Y-27632 is a selective inhibitor of ROCKs including p160ROCK (Ki: 140 nM) and ROCK2 (IC50: 800 nM).
Biological Applications: Y-27632HCl, as a ROCK inhibitor, has significant applications not only in diseases like cancer but also in organoid culture and stem cell research, contributing to the maintenance of cell survival and functionality, thus holding crucial value in various fields. Common applications:
- Cryopreservation of stem cells: Y-27632 helps prevent dissociation-related cell apoptosis during the low-temperature preservation of stem cells and enhances cell viability post-thaw.
- Pancreatic ductal adenocarcinoma organoid culture: Y-27632 serves as a supplement to the culture medium.
- Mouse embryonic stem cells: Y-27632 inhibits the Rho kinase (Rho) in these cells.
- Human embryonic stem cells and induced pluripotent stem cells (iPSCs): Y-27632 inhibits Rho-associated protein kinase (ROCK).
These diverse applications highlight the versatility and importance of Y-27632HCl in various research settings, promising advancements in cell biology and regenerative medicine.
Research Area: Cancer, glaucoma, asthma, erectile dysfunction, insulin resistance, neurodegeneration, osteoporosis, renal failure, fibrosis, and graft-versus-host disease.
Target: ROCK; Apoptosis
Z-VAD-FMK
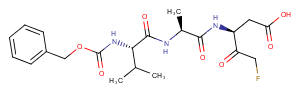
Z-VAD-FMK (Caspase Inhibitor VI) is an irreversible pan-caspase inhibitor.
Bioactivity: Z-VAD-FMK (Caspase Inhibitor VI) is an irreversible pan-caspase inhibitor.
Biological Applications: Z-VAD-FMK, as a broad-spectrum inhibitor of Caspase, plays a crucial role in apoptosis research. Numerous preclinical studies, both in vitro and in vivo, indicate that Caspase primarily acts as an inflammatory and apoptotic mediator in various pathologies. Consequently, several Caspase inhibitors have been patented for their anti-inflammatory and apoptotic functions. However, due to factors such as drug toxicity, their application is currently limited to preclinical research. Although some studies propose novel therapeutic approaches using nanoparticle delivery systems and CRISPR/Cas9 gene editing to improve drug delivery and reduce drug-induced toxicity, targeting individual Caspases separately, these remain short-term solutions. Because the lack of Caspase activity can increase the crosstalk between cell death and inflammatory pathways, there are concerns about the long-term efficacy of Caspase inhibitors. If inhibitors increase the risk of cell death and inflammatory reactions, they may exacerbate diseases. Therefore, it is crucial to clearly determine the specific mechanisms of action of Caspase inhibitors in preclinical models.
Mechanism of Action: Z-VAD (OH)-FMK is a carboxylic acid compound that can be directly added to cell culture media without the need for esterase pre-treatment.
Research Area: Neurological disorders (epilepsy, Parkinson’s, etc.), Inflammatory diseases (rheumatoid arthritis, sepsis), AIDS, Obesity, diabetes, Liver disease, Breast cancer, lung cancer, etc.
Target: Caspase
Z-VAD(OMe)-FMK
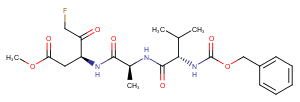
Z-VAD(OMe)-FMK is a cell-permeable and irreversible pan-caspase inhibitor. It inhibits cleavage of PARP, preventing apoptosis when used at 10-50 uM.
Bioactivity: Z-VAD(OMe)-FMK is a cell-permeable and irreversible pan-caspase inhibitor. It inhibits cleavage of PARP, preventing apoptosis when used at 10-50 μM.
Biological Applications: Z-VAD-FMK, as a broad-spectrum inhibitor of Caspase, plays a crucial role in apoptosis research. Numerous preclinical studies, both in vitro and in vivo, indicate that Caspase primarily acts as an inflammatory and apoptotic mediator in various pathologies. Consequently, several Caspase inhibitors have been patented for their anti-inflammatory and apoptotic functions. However, due to factors such as drug toxicity, their application is currently limited to preclinical research. Although some studies propose novel therapeutic approaches using nanoparticle delivery systems and CRISPR/Cas9 gene editing to improve drug delivery and reduce drug-induced toxicity, targeting individual Caspases separately, these remain short-term solutions. Because the lack of Caspase activity can increase the crosstalk between cell death and inflammatory pathways, there are concerns about the long-term efficacy of Caspase inhibitors. If inhibitors increase the risk of cell death and inflammatory reactions, they may exacerbate diseases. Therefore, it is crucial to clearly determine the specific mechanisms of action of Caspase inhibitors in preclinical models.
Mechanism of Action: Z-VAD (OMe)-FMK is a cell-permeable, irreversible caspase inhibitor that can inhibit caspases and apoptosis in tumour cells in vitro. The methyl ester compound undergoes hydrolysis by endogenous esterase activity upon entering the cell, generating the biologically active form. Therefore, pre-treatment with esterase is required when using it with isolated, purified, or recombinant caspases.
Research Area: Neurological disorders (epilepsy, Parkinson’s, etc.), Inflammatory diseases (rheumatoid arthritis, sepsis), AIDS, Obesity, diabetes, Liver disease, Breast cancer, lung cancer, etc.
Target: Caspase
Information provided by TargetMol.
Caltag Medsystems is the distributor of TargetMol products in the UK and Ireland. If you have any questions about these products, please contact us.